Plot Dual-Endpoint Design Simulation Summary
Source:R/Simulations-methods.R
plot-DualSimulationsSummary-missing-method.Rd![[Stable]](figures/lifecycle-stable.svg)
Graphical display of dual-endpoint simulation summary.
This plot method can be applied to DualSimulationsSummary objects in
order to summarize them graphically. Possible type of plots at the moment
are those listed in plot,SimulationsSummary,missing-method plus:
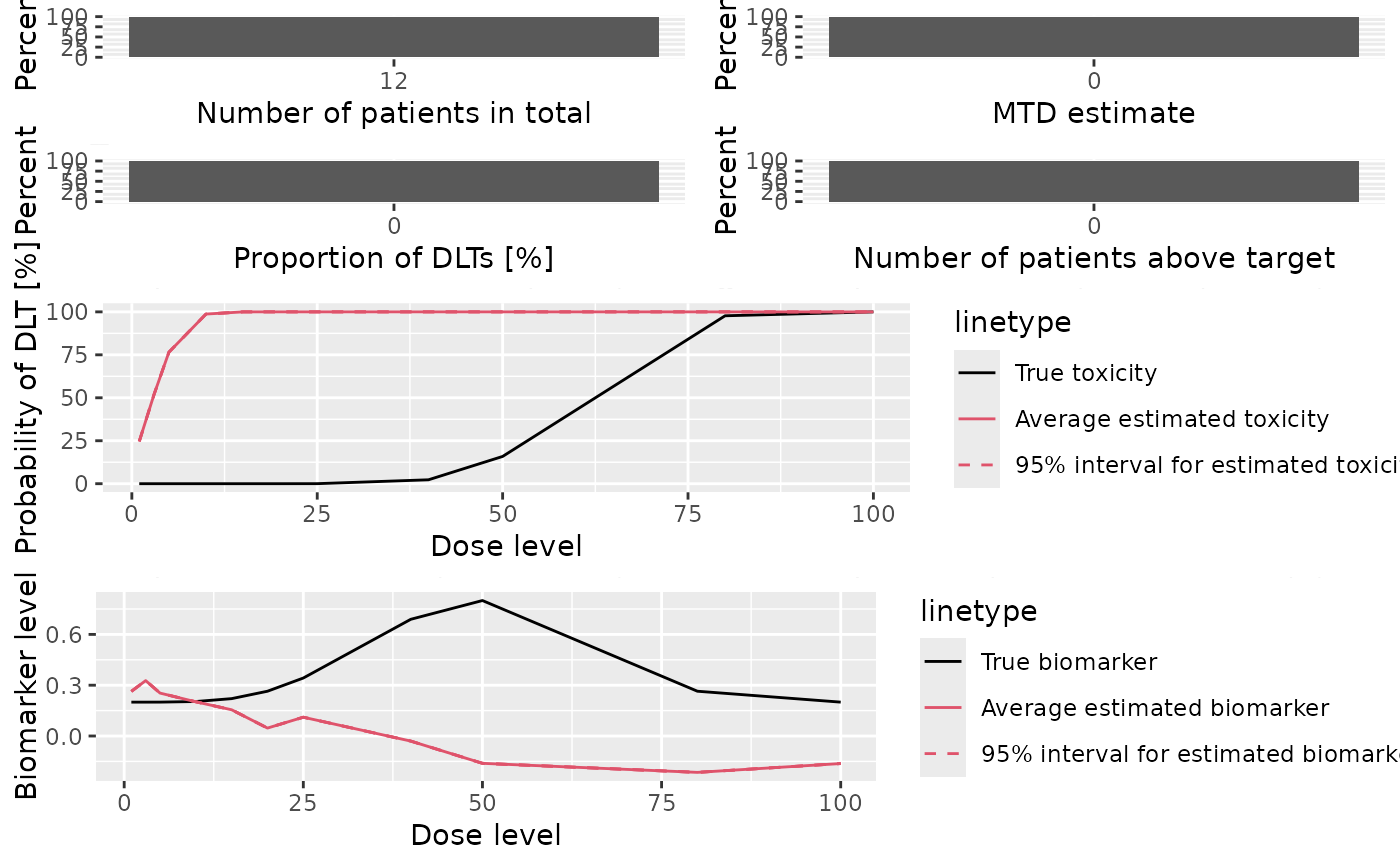
- meanBiomarkerFit
Plot showing the average fitted dose-biomarker curve across the trials, together with 95% credible intervals, and comparison with the assumed truth (as specified by the
trueBiomarkerargument tosummary,DualSimulations-method)
You can specify any subset of these in the type argument.
Examples
# Define the dose-grid.
emptydata <- DataDual(doseGrid = c(1, 3, 5, 10, 15, 20, 25, 40, 50, 80, 100))
# Initialize the CRM model.
my_model <- DualEndpointRW(
mean = c(0, 1),
cov = matrix(c(1, 0, 0, 1), nrow = 2),
sigma2betaW = 0.01,
sigma2W = c(a = 0.1, b = 0.1),
rho = c(a = 1, b = 1),
rw1 = TRUE
)
# Choose the rule for selecting the next dose.
my_next_best <- NextBestDualEndpoint(
target = c(0.9, 1),
overdose = c(0.35, 1),
max_overdose_prob = 0.25
)
# Choose the rule for the cohort-size.
my_size1 <- CohortSizeRange(
intervals = c(0, 30),
cohort_size = c(1, 3)
)
my_size2 <- CohortSizeDLT(
intervals = c(0, 1),
cohort_size = c(1, 3)
)
my_size <- maxSize(my_size1, my_size2)
# Choose the rule for stopping.
my_stopping4 <- StoppingTargetBiomarker(
target = c(0.9, 1),
prob = 0.5
)
# Only 10 patients here for illustration!
my_stopping <- my_stopping4 | StoppingMinPatients(10) | StoppingMissingDose()
# Choose the rule for dose increments.
my_increments <- IncrementsRelative(
intervals = c(0, 20),
increments = c(1, 0.33)
)
# Initialize the design.
my_design <- DualDesign(
model = my_model,
data = emptydata,
nextBest = my_next_best,
stopping = my_stopping,
increments = my_increments,
cohort_size = CohortSizeConst(3),
startingDose = 3
)
# Define scenarios for the TRUE toxicity and efficacy profiles.
beta_mod <- function(dose, e0, eMax, delta1, delta2, scal) {
maxDens <- (delta1^delta1) *
(delta2^delta2) /
((delta1 + delta2)^(delta1 + delta2))
dose <- dose / scal
e0 + eMax / maxDens * (dose^delta1) * (1 - dose)^delta2
}
true_biomarker <- function(dose) {
beta_mod(
dose,
e0 = 0.2,
eMax = 0.6,
delta1 = 5,
delta2 = 5 * 0.5 / 0.5,
scal = 100
)
}
true_tox <- function(dose) {
pnorm((dose - 60) / 10)
}
# Draw the TRUE profiles.
par(mfrow = c(1, 2))
curve(true_tox(x), from = 0, to = 80)
curve(true_biomarker(x), from = 0, to = 80)
# Run the simulation on the desired design.
# We only generate 1 trial outcome here for illustration, for the actual study.
# For illustration purpose we will use 5 burn-ins to generate 20 samples,
# this should be increased of course.
my_sims <- simulate(
object = my_design,
trueTox = true_tox,
trueBiomarker = true_biomarker,
sigma2W = 0.01,
rho = 0,
nsim = 1,
parallel = FALSE,
seed = 3,
startingDose = 6,
mcmcOptions = McmcOptions(
burnin = 5,
step = 1,
samples = 20
)
)
# Plot the summary of the Simulations.
plot(summary(my_sims, trueTox = true_tox, trueBiomarker = true_biomarker))